This was a project I worked on as part of the BioXFEL summer internship program. I worked with Dr. Sarah Bowman and other members of Hauptman-Woodward Medical Research Institute (HWI).
If you are short on time and don’t want to read these entire post here are the important links.
So what is “high-throughput crystallography”?
Lets say you are a researcher, trying to develop a treatment for a novel virus that is quickly spreading across the globe. You know that the binding between the viruses outer spike protein and a receptor on the cells of lung tissue is critical to the pathogens virulence.
If you knew that spike protein looked like, at the atomic, level you could more easily design a small-molecule drug that would interrupt its “normal” interaction with the lung tissue receptor protein. Of course it is non-trivial to actually “look” at a protein. In fact, the wavelength of visible light is much larger than the atoms you are trying to peak at.
To get around this problem a number of specialized techniques have been developed, the most commonly employed being X-ray crystallography (although keep your eye on emerging cryo-EM technologies.)
Just like minerals in the Earth, proteins can form crystalize, organizing into an infinitely repeating, regular pattern of molecules.

Protein crystals grown in space. NASA Marshall Space Flight Center (NASA-MSFC) - NASA
To grossly oversimplify, X-ray crystallography takes advantage of the small wavelength of X-rays and the repeating structure of protein crystals to “amplify” the extremely small signal produced by a single molecule allowing researchers, along with the help of a lot of fast-fourier transforms to piece together the 3D structure of a protein with resolutions below 1.5 Å (angstroms).
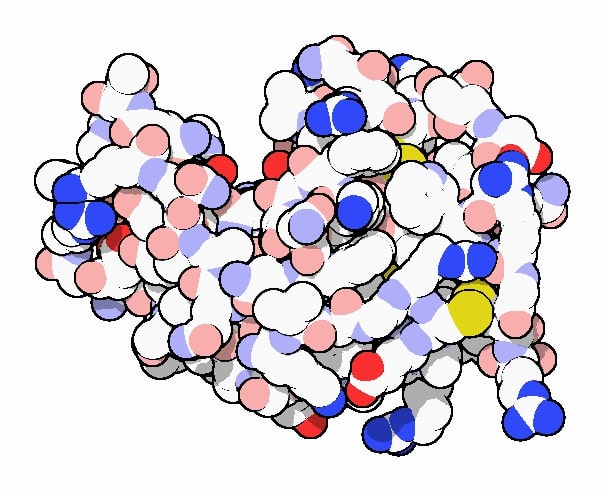
Crystal structure of lysozyme as rendered by David Goodsell doi:10.2210/rcsb_pdb/mom_2000_9
The only problem is that actually growing these protein crystals is extremely challenging. There is currently no reliable way to predict the range of chemical conditions that might result in a successful crystallization due to the relatively chaotic nature of crystal growth. This often means even “homology” based approaches (protein X which is similar to protein Y so conditions for protein X should work for Y) often fail. Overall, this often makes growing protein crystals a labor intensive ordeal with with no guarantee of success.
In most cases this will involve researchers manually creating grids of chemical conditions mixed with different concentrations of protein in the hopes that one produces crystals. Additionally, these types of manual screens require a large amount of extremely pure protein which is often difficult and or expensive to obtain.
High-throughput crystallography utilizes liquid handling robotics, automated microscopy, and nano-liter well sizes in order to do effectively the same process while screening more chemical conditions and using less protein. This makes it appealing for researchers working with many or difficult to crystalize proteins.
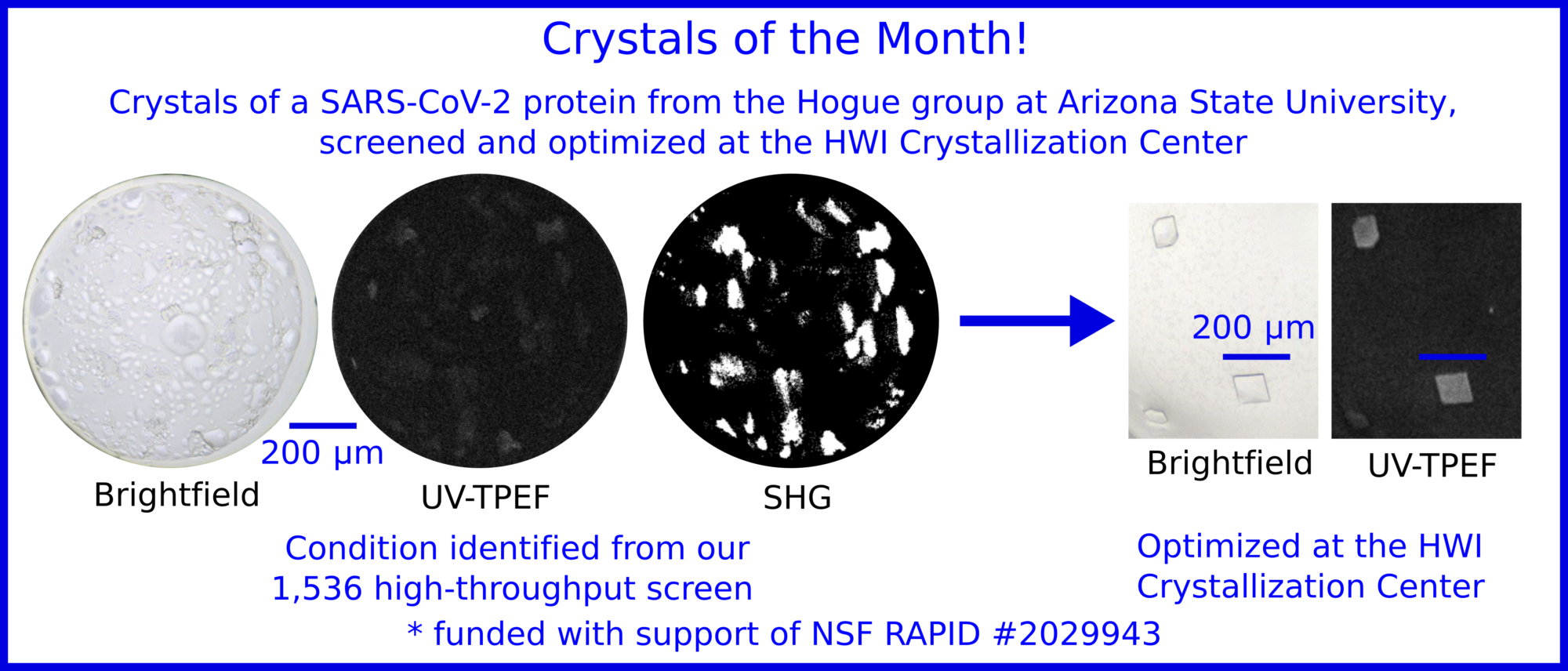
SARS-CoV-2 protein crystals grown at Hauptmann-Woodward Medical Research Institute High-Throughput Crystallization Center
More data more problems: Polo’s motivation
The number of conditions that high-throughput approaches is at the same time its selling point and greatest challenge. A single-sample screening run can produce over 15360 images that have to been sorted though to determine if any contain crystals. This is the worst king of monotonous task as it is highly repetitive but yet required acute concentration, as missing a crystal in one image could mean the difference between a successful and failed study.
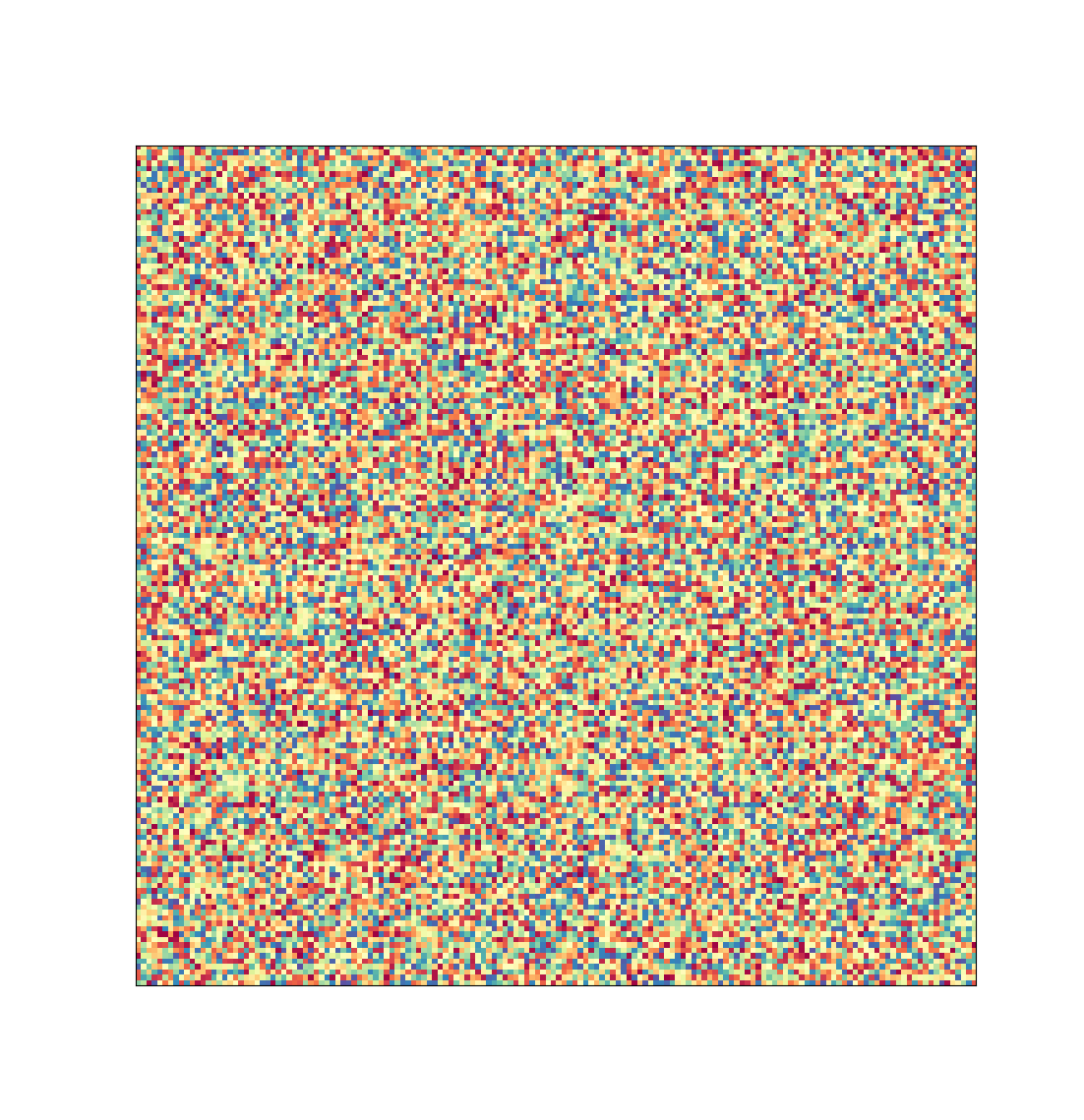
Representation of the image data that would be created by a typical high-throughput screen of one protein sample at HWI. Each pixel represents one image a researcher would have to manually review.
Fortunately, advances in machine learning have allowed for the development of models that can do this work automatically and with reasonable accuracy. The most recent iteration of this has been the MARCO (Machine Recognition of Crystallization Outcomes) model published by Andrew Bruno et al in 2018.
The MARCO model opened the door for experts to automatically classify crystallization images, but lack of a graphical interface and data management program made it largely inaccessibly to users without Python and or command line experience. Therefore the goal of my project was to produce a free, easy to use graphical interface for both HWI and non-HWI users that would integrate the MARCO model with the best features of existing image review software.
Outcome
Despite the short development timeline (largely 12 weeks over the summer) I would say Polo was a success. Over that period of time I was able to release versions of the program for Linux, Mac and Windows, created a website to host the documentation (which you can view here) and began drafting a manuscript which is currently under review by the Journal of Applied Crystallography.
Here are a few images of some of the Polo interfaces, for full details, user-guides, code documentation and video tutorials please visit the Polo website.
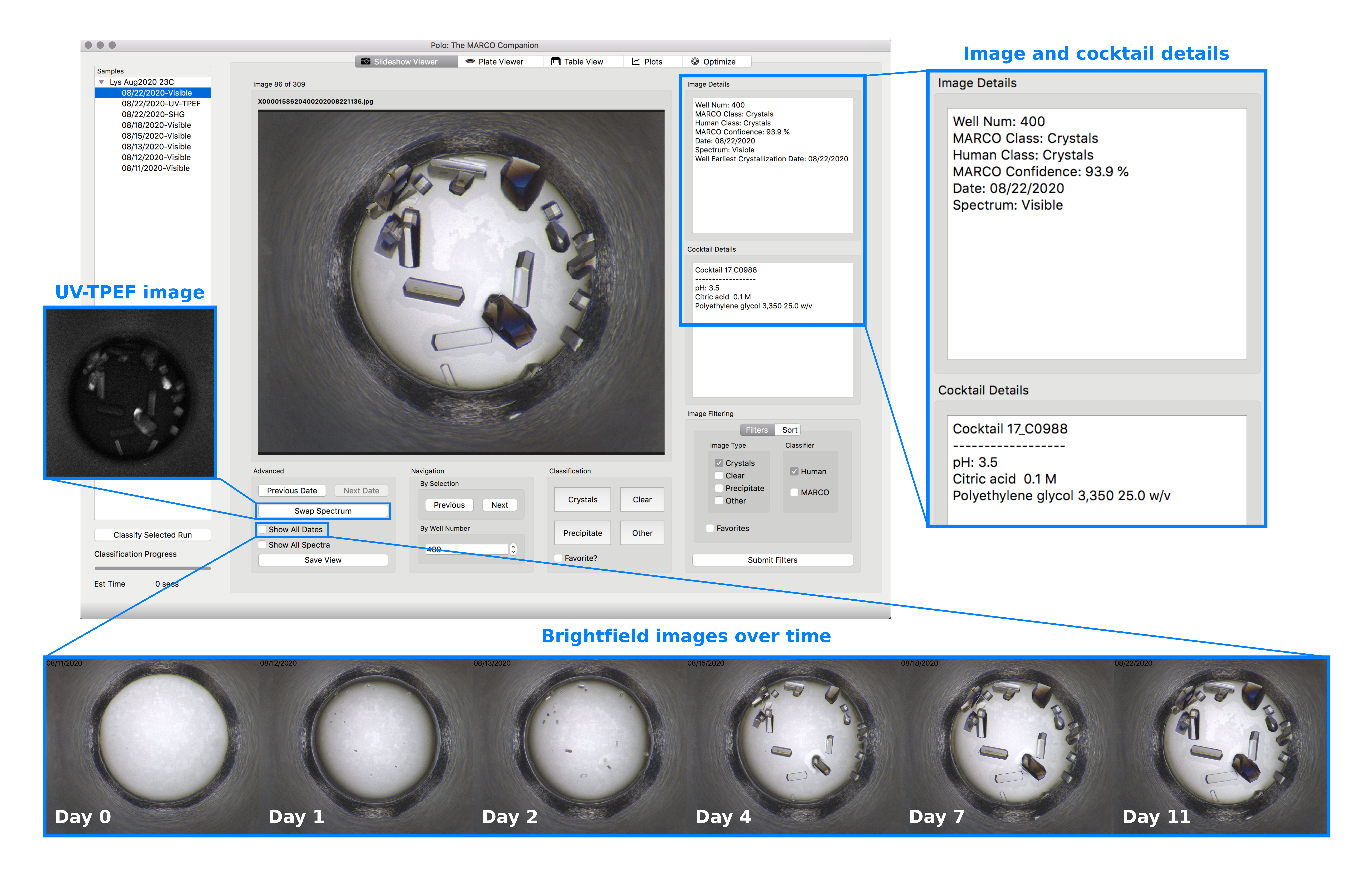
Composited image of different viewing options of the Polo slideshow viewer interface
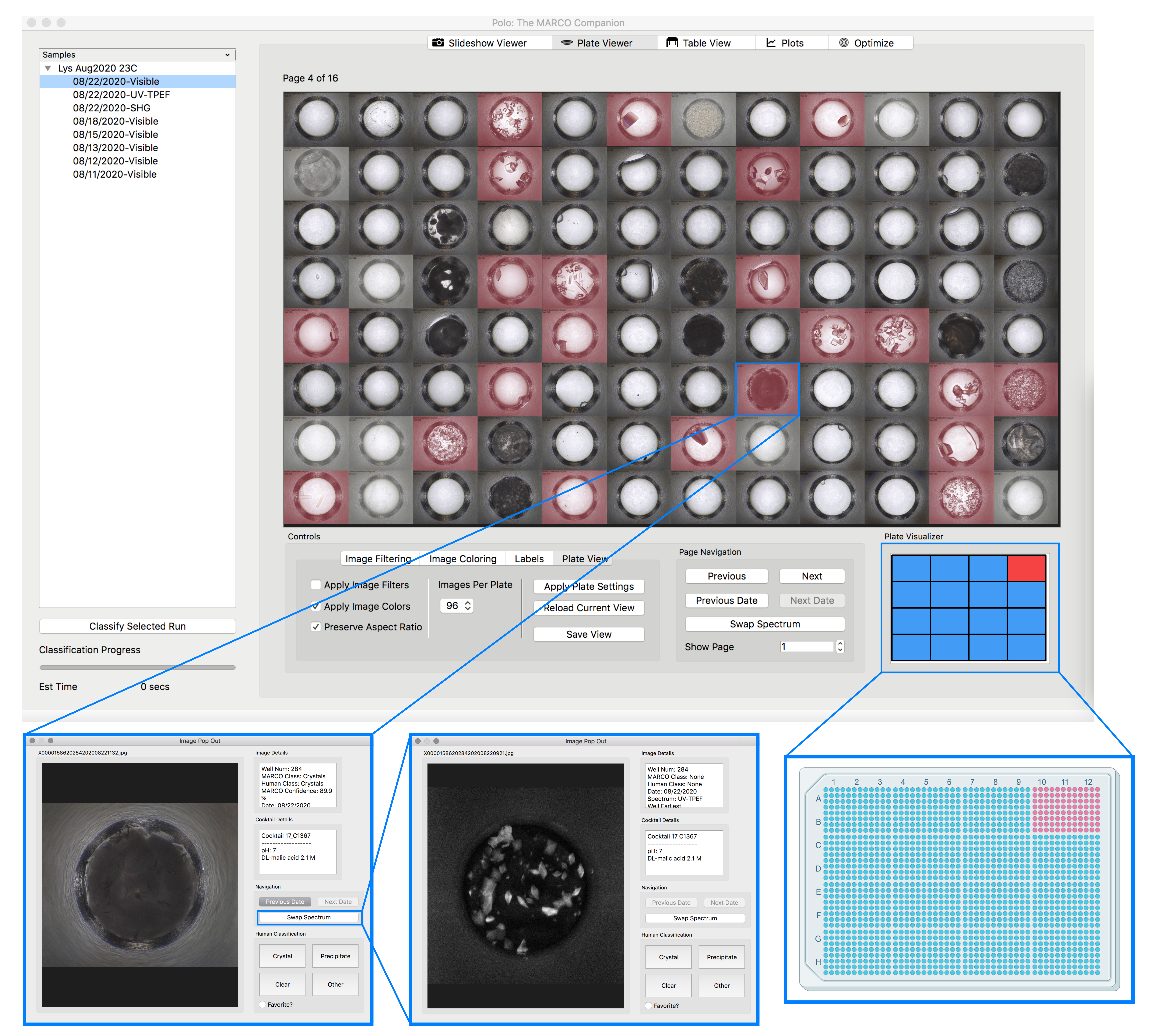
Composited image of different viewing options of the Polo plate viewer interface
The most recent version of Polo can be downloaded from this link. Beyond automatic image classification via MARCO, Polo includes a number of useful tools and interfaces. HWI and non-HWI users alike can
- Download images directly from Polo using the integrated FTP client
- Export their results to PowerPoint presentations, csv or json files
- View images as slideshows or in arrays of up to 96 images
- Easily associate visible light microscopy images to alternative spectrums such as UV-TPEF and quickly swap between the two
Polo is also fully open source and free for academic and commercial use!
There are also a number of resources for getting starting with Polo, including video guides where I walk you through using Polo’s main interfaces, written tutorials and API style code documentation for anyone interesting in extending or modifying Polo.
Future goals and collaboration
My personal goal for Polo was to create something that was accessible, easy to use and significantly reduced human work. If, after trying out the program or reviewing any of the links above you have suggestion on how Polo can better fulfil those goals please let me know! Feel free to reach out through GitHub, pull request, or through the Polo bug and suggestion Google form.
I still work to maintain Polo and do my best to respond to questions and fix bugs reported by users. If any of the above is interesting to you or you have questions about how Polo might be able to help you, your lab or organization please reach out! I would love to talk.
In the future I would like involve researchers from other high-throughout facilities to extend the functionality and flexibility of Polo.